Part 2: Hello GATK¶
The GATK (Genome Analysis Toolkit) is a widely used software package developed by the Broad Institute to analyze high-throughput sequencing data. We're going to use GATK and a related tool, Samtools, in a very basic pipeline that identifies genomic variants through a method called variant calling.
Note
Don't worry if you're not familiar with GATK or genomics in general. We'll summarize the necessary concepts as we go, and the workflow implementation principles we demonstrate here apply broadly to any command line tool that takes in some input files and produce some output files.
A full variant calling pipeline typically involves a lot of steps. For simplicity, we are only going to look at the core variant calling steps.
Method overview¶
- Generate an index file for each BAM input file using Samtools
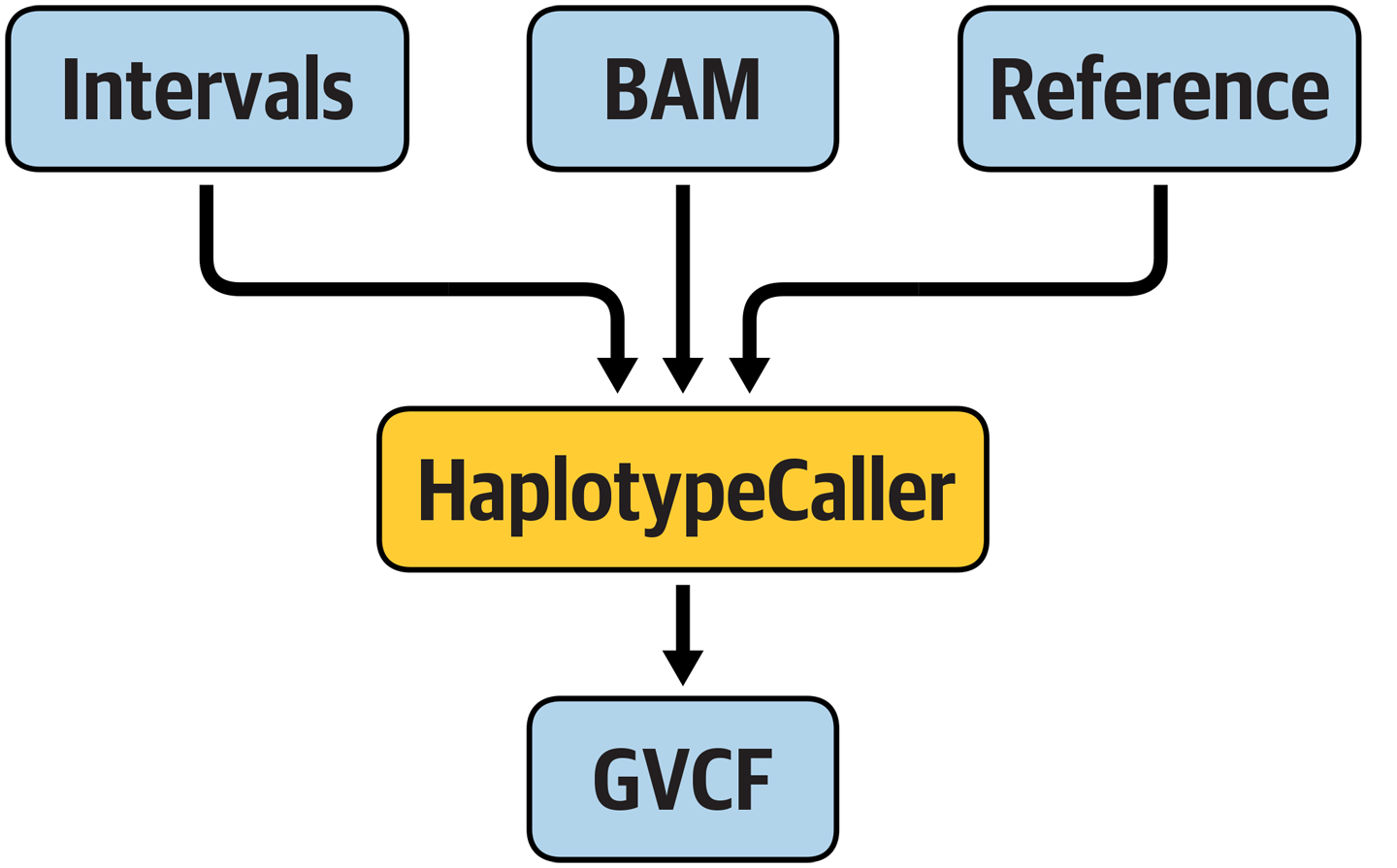
- Run the GATK HaplotypeCaller on each BAM input file to generate per-sample variant calls in GVCF (Genomic Variant Call Format)
Dataset¶
- A reference genome consisting of the human chromosome 20 (from hg19/b37) and its accessory files (index and sequence dictionary). The reference files are compressed to keep the Gitpod size small so we'll have to decompress them in order to use them.
- Three whole genome sequencing samples corresponding to a family trio (mother, father and son), which have been subset to a small portion on chromosome 20 to keep the file sizes small. The sequencing data is in BAM (Binary Alignment Map) format, i.e. genome sequencing reads that have already been mapped to the reference genome.
- A list of genomic intervals, i.e. coordinates on the genome where our samples have data suitable for calling variants.
0. Warmup: Run Samtools and GATK directly¶
Just like in the Hello World example, we want to try out the commands manually before we attempt to wrap them in a workflow. The difference here is that we're going to use Docker containers to obtain and run the tools.
0.1. Index a BAM input file with Samtools¶
0.1.1. Pull the samtools container¶
0.1.2. Spin up the container interactively¶
0.1.3. Run the indexing command¶
0.1.4. Check that the BAM index has been produced¶
This should show:
Where reads_mother.bam.bai has been created as an index to reads_mother.bam.
0.1.5. Exit the container¶
0.2. Call variants with GATK HaplotypeCaller¶
0.2.1. Decompress the reference genome files¶
0.2.2. Pull the GATK container¶
0.2.3. Spin up the container interactively¶
0.2.4. Run the variant calling command¶
gatk HaplotypeCaller \
-R /data/ref/ref.fasta \
-I /data/bam/reads_mother.bam \
-O reads_mother.g.vcf \
-L /data/intervals.list \
-ERC GVCF
0.2.5. Check the contents of the output file¶
1. Write a single-stage workflow that runs Samtools index on a BAM file¶
1.1. Define the indexing process¶
/*
* Generate BAM index file
*/
process SAMTOOLS_INDEX {
container 'quay.io/biocontainers/samtools:1.19.2--h50ea8bc_1'
input:
path input_bam
output:
path "${input_bam}.bai"
"""
samtools index '$input_bam'
"""
}
1.2. Add parameter declarations up top¶
/*
* Pipeline parameters
*/
// Execution environment setup
params.baseDir = "/workspace/gitpod/hello-nextflow"
$baseDir = params.baseDir
// Primary input
params.reads_bam = "${baseDir}/data/bam/reads_mother.bam"
1.3. Add workflow block to run SAMTOOLS_INDEX¶
workflow {
// Create input channel (single file via CLI parameter)
reads_ch = Channel.from(params.reads_bam)
// Create index file for input BAM file
SAMTOOLS_INDEX(reads_ch)
}
1.4. Run it to verify you can run the indexing step¶
Should produce something like:
N E X T F L O W ~ version 23.10.1
Launching `hello-gatk.nf` [compassionate_cray] DSL2 - revision: 9b97744397
executor > local (1)
[bf/072bd7] process > SAMTOOLS_INDEX (1) [100%] 1 of 1 ✔
Takeaway¶
You know how to wrap a real bioinformatics tool in a single-step Nextflow workflow.
What's next?¶
Add a second step that consumes the output of the first.
2. Add a second step that runs GATK HaplotypeCaller on the indexed BAM file¶
2.1. Define the variant calling process¶
/*
* Call variants with GATK HapolotypeCaller in GVCF mode
*/
process GATK_HAPLOTYPECALLER {
container "broadinstitute/gatk:4.5.0.0"
input:
path input_bam
path input_bam_index
path ref_fasta
path ref_index
path ref_dict
path interval_list
output:
path "${input_bam}.g.vcf"
path "${input_bam}.g.vcf.idx"
"""
gatk HaplotypeCaller \
-R ${ref_fasta} \
-I ${input_bam} \
-O ${input_bam}.g.vcf \
-L ${interval_list} \
-ERC GVCF
"""
}
2.2. Add accessory inputs up top¶
// Accessory files
params.genome_reference = "${baseDir}/data/ref/ref.fasta"
params.genome_reference_index = "${baseDir}/data/ref/ref.fasta.fai"
params.genome_reference_dict = "${baseDir}/data/ref/ref.dict"
params.calling_intervals = "${baseDir}/data/intervals.list"
2.3. Add a call to the workflow block to run GATK_HAPLOTYPECALLER¶
// Call variants from the indexed BAM file
GATK_HAPLOTYPECALLER(
reads_ch,
SAMTOOLS_INDEX.out,
params.genome_reference,
params.genome_reference_index,
params.genome_reference_dict,
params.calling_intervals
)
2.4. Run the workflow to verify that the variant calling step works¶
Now we see the two processes being run:
N E X T F L O W ~ version 23.10.1
Launching `hello-gatk.nf` [lethal_keller] DSL2 - revision: 30a64b9325
executor > local (2)
[97/0f85bf] process > SAMTOOLS_INDEX (1) [100%] 1 of 1 ✔
[2d/43c247] process > GATK_HAPLOTYPECALLER (1) [100%] 1 of 1 ✔
If you check the work directory, you'll find the output file reads_mother.bam.g.vcf. Because this is a small test file, you can click on it to open it and view the contents, which consist of 92 lines of header metadata followed by a list of genomic variant calls, one per line.
Note
A GVCF is a special kind of VCF that contains non-variant records as well as variant calls. The first actual variant call in this file occurs at line 325:
Takeaway¶
You know how to make a very basic two-step variant calling workflow.
What's next?¶
Make the workflow handle multiple samples in bulk.
3. Adapt the workflow to run on a batch of samples¶
3.1. Turn the input param declaration into a list of the three samples¶
// Primary input
params.reads_bam = ["${baseDir}/data/gatk/bam/reads_mother.bam",
"${baseDir}/data/gatk/bam/reads_father.bam",
"${baseDir}/data/gatk/bam/reads_son.bam"]
3.2. Run the workflow to verify that it runs on all three samples¶
Uh-oh! Sometimes it works, but sometimes, some of the runs fail with an error like this:
executor > local (6) [f3/80670d] process > SAMTOOLS_INDEX (1) [100%] 3 of 3 ✔ [27/78b83d] process > GATK_HAPLOTYPECALLER (3) [100%] 1 of 1, failed: 1 ERROR ~ Error executing process > 'GATK_HAPLOTYPECALLER (1)'
Caused by: Process
GATK_HAPLOTYPECALLER (1)terminated with an error exit status (2)Command executed:
gatk HaplotypeCaller -R ref.fasta -I reads_mother.bam -O reads_mother.bam.g.vcf -L intervals-min.list -ERC GVCF
Command exit status: 2
Command output: (empty)
Command error: 04:52:05.954 INFO HaplotypeCaller - Java runtime: OpenJDK 64-Bit Server VM v17.0.9+9-Ubuntu-122.04 04:52:05.955 INFO HaplotypeCaller - Start Date/Time: March 15, 2024 at 4:52:05 AM GMT 04:52:05.955 INFO HaplotypeCaller - ------------------------------------------------------------ 04:52:05.955 INFO HaplotypeCaller - ------------------------------------------------------------ 04:52:05.956 INFO HaplotypeCaller - HTSJDK Version: 4.1.0 04:52:05.956 INFO HaplotypeCaller - Picard Version: 3.1.1 04:52:05.956 INFO HaplotypeCaller - Built for Spark Version: 3.5.0 04:52:05.957 INFO HaplotypeCaller - HTSJDK Defaults.COMPRESSION_LEVEL : 2 04:52:05.957 INFO HaplotypeCaller - HTSJDK Defaults.USE_ASYNC_IO_READ_FOR_SAMTOOLS : false 04:52:05.957 INFO HaplotypeCaller - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_SAMTOOLS : true 04:52:05.957 INFO HaplotypeCaller - HTSJDK Defaults.USE_ASYNC_IO_WRITE_FOR_TRIBBLE : false 04:52:05.958 INFO HaplotypeCaller - Deflater: IntelDeflater 04:52:05.958 INFO HaplotypeCaller - Inflater: IntelInflater 04:52:05.958 INFO HaplotypeCaller - GCS max retries/reopens: 20 04:52:05.958 INFO HaplotypeCaller - Requester pays: disabled 04:52:05.959 INFO HaplotypeCaller - Initializing engine 04:52:06.563 INFO IntervalArgumentCollection - Processing 20000 bp from intervals 04:52:06.572 INFO HaplotypeCaller - Done initializing engine 04:52:06.575 INFO HaplotypeCallerEngine - Tool is in reference confidence mode and the annotation, the following changes will be made to any specified annotations: 'StrandBiasBySample' will be enabled. 'ChromosomeCounts', 'FisherStrand', 'StrandOddsRatio' and 'QualByDepth' annotations have been disabled 04:52:06.653 INFO NativeLibraryLoader - Loading libgkl_utils.so from jar:file:/gatk/gatk-package-4.5.0.0-local.jar!/com/intel/gkl/native/libgkl_utils.so 04:52:06.656 INFO NativeLibraryLoader - Loading libgkl_smithwaterman.so from jar:file:/gatk/gatk-package-4.5.0.0-local.jar!/com/intel/gkl/native/libgkl_smithwaterman.so 04:52:06.657 INFO SmithWatermanAligner - Using AVX accelerated SmithWaterman implementation 04:52:06.662 INFO HaplotypeCallerEngine - Standard Emitting and Calling confidence set to -0.0 for reference-model confidence output 04:52:06.663 INFO HaplotypeCallerEngine - All sites annotated with PLs forced to true for reference-model confidence output 04:52:06.676 INFO NativeLibraryLoader - Loading libgkl_pairhmm_omp.so from jar:file:/gatk/gatk-package-4.5.0.0-local.jar!/com/intel/gkl/native/libgkl_pairhmm_omp.so 04:52:06.756 INFO IntelPairHmm - Flush-to-zero (FTZ) is enabled when running PairHMM 04:52:06.757 INFO IntelPairHmm - Available threads: 16 04:52:06.757 INFO IntelPairHmm - Requested threads: 4 04:52:06.757 INFO PairHMM - Using the OpenMP multi-threaded AVX-accelerated native PairHMM implementation 04:52:06.954 INFO ProgressMeter - Starting traversal 04:52:06.955 INFO ProgressMeter - Current Locus Elapsed Minutes Regions Processed Regions/Minute 04:52:06.967 INFO VectorLoglessPairHMM - Time spent in setup for JNI call : 0.0 04:52:06.968 INFO PairHMM - Total compute time in PairHMM computeLogLikelihoods() : 0.0 04:52:06.969 INFO SmithWatermanAligner - Total compute time in native Smith-Waterman : 0.00 sec 04:52:06.971 INFO HaplotypeCaller - Shutting down engine [March 15, 2024 at 4:52:06 AM GMT] org.broadinstitute.hellbender.tools.walkers.haplotypecaller.HaplotypeCaller done. Elapsed time: 0.03 minutes. Runtime.totalMemory()=629145600
A USER ERROR has occurred: Traversal by intervals was requested but some input files are not indexed. Please index all input files:
samtools index reads_mother.bam
Set the system property GATK_STACKTRACE_ON_USER_EXCEPTION (--java-options '-DGATK_STACKTRACE_ON_USER_EXCEPTION=true') to print the stack trace. Using GATK jar /gatk/gatk-package-4.5.0.0-local.jar Running: java -Dsamjdk.use_async_io_read_samtools=false -Dsamjdk.use_async_io_write_samtools=true -Dsamjdk.use_async_io_write_tribble=false -Dsamjdk.compression_level=2 -jar /gatk/gatk-package-4.5.0.0-local.jar HaplotypeCaller -R ref.fasta -I reads_mother.bam -O reads_mother.bam.g.vcf -L intervals-min.list -ERC GVCF
Work dir: /workspace/gitpod/nf-training/work/22/611b8c5703daaf459188d79cd68db0
Tip: you can try to figure out what's wrong by changing to the process work dir and showing the script file named
.command.sh-- Check '.nextflow.log' file for details
Why does this happen? Because the order of outputs is not guaranteed, so the script as written so far is not safe for running on multiple samples!
3.3. Change the output of the SAMTOOLS_INDEX process into a tuple that keeps the input file and its index together¶
Before:
After:
3.4. Change the input to the GATK_HAPLOTYPECALLER process to be a tuple¶
Before:
After:
3.5. Update the call to GATK_HAPLOTYPECALLER in the workflow block¶
Before:
After:
3.6. Run the workflow to verify it works correctly on all three samples now¶
This time everything should run correctly:
N E X T F L O W ~ version 23.10.1
Launching `hello-gatk.nf` [adoring_hopper] DSL2 - revision: 8cad21ea51
[e0/bbd6ef] Submitted process > SAMTOOLS_INDEX (3)
[71/d26b2c] Submitted process > SAMTOOLS_INDEX (2)
[e6/6cad6d] Submitted process > SAMTOOLS_INDEX (1)
[26/73dac1] Submitted process > GATK_HAPLOTYPECALLER (1)
[23/12ed10] Submitted process > GATK_HAPLOTYPECALLER (2)
[be/c4a067] Submitted process > GATK_HAPLOTYPECALLER (3)
Takeaway¶
You know how to make a variant calling workflow run on multiple samples (independently).
What's next?¶
Make it easier to handle samples in bulk.
4. Make it nicer to run on arbitrary samples by using a list of files as input¶
4.1. Create a text file listing the input paths¶
sample_bams.txt:
/workspace/gitpod/hello-nextflow/data/bam/reads_mother.bam
/workspace/gitpod/hello-nextflow/data/bam/reads_father.bam
/workspace/gitpod/hello-nextflow/data/bam/reads_son.bam
4.2. Update the parameter default¶
Before:
// Primary input
params.reads_bam = ["${baseDir}/data/bam/reads_mother.bam",
"${baseDir}/data/bam/reads_father.bam",
"${baseDir}/data/bam/reads_son.bam"]
After:
// Primary input (list of input files, one per line)
params.reads_bam = "${baseDir}/data/bam/sample_bams.txt"
4.3. Update the channel factory to read lines from a file¶
Before:
After:
// Create input channel from list of input files in plain text
reads_ch = Channel.fromPath(params.reads_bam).splitText
4.4. Run the workflow to verify that it works correctly¶
This should produce essentially the same result as before:
N E X T F L O W ~ version 23.10.1
Launching `hello-gatk.nf` [kickass_faggin] DSL2 - revision: dcfa9f34e3
[ff/0c08e6] Submitted process > SAMTOOLS_INDEX (2)
[75/bcae76] Submitted process > SAMTOOLS_INDEX (1)
[df/75d25a] Submitted process > SAMTOOLS_INDEX (3)
[00/295d75] Submitted process > GATK_HAPLOTYPECALLER (1)
[06/89c1d1] Submitted process > GATK_HAPLOTYPECALLER (2)
[58/866482] Submitted process > GATK_HAPLOTYPECALLER (3)
Takeaway¶
You know how to make a variant calling workflow handle a list of input samples.
What's next?¶
Turn the list of input files into a samplesheet by including some metadata.
5. Upgrade to using a (primitive) samplesheet¶
This is a very common pattern in Nextflow pipelines.
5.1. Add a header line and the sample IDs to a copy of the sample list, in CSV format¶
samplesheet.csv:
ID,reads_bam
NA12878,/workspace/gitpod/hello-nextflow/data/bam/reads_mother.bam
NA12877,/workspace/gitpod/hello-nextflow/data/bam/reads_father.bam
NA12882,/workspace/gitpod/hello-nextflow/data/bam/reads_son.bam
5.2. Update the parameter default¶
Before:
// Primary input (list of input files, one sample per line)
params.reads_bam = "${baseDir}/data/bam/sample_bams.txt"
After:
// Primary input (samplesheet in CSV format with ID and file path, one sample per line)
params.reads_bam = "${baseDir}/data/samplesheet.csv"
5.3. Update the channel factory to parse a CSV file¶
Before:
// Create input channel from list of input files in plain text
reads_ch = Channel.fromPath(params.reads_bam).splitText()
After:
// Create input channel from samplesheet in CSV format
reads_ch = Channel.fromPath(params.reads_bam)
.splitCsv(header: true)
.map{row -> [row.id, file(row.reads_bam)]}
5.4. Add the sample ID to the SAMTOOLS_INDEX input definition¶
Before:
After:
5.5. Run the workflow to verify that it works¶
If everything is wired up correctly, it should produce essentially the same result.
N E X T F L O W ~ version 23.10.1
Launching `hello-gatk.nf` [extravagant_panini] DSL2 - revision: 56accbf948
[19/00f4a5] Submitted process > SAMTOOLS_INDEX (3)
[4d/532d60] Submitted process > SAMTOOLS_INDEX (1)
[08/5628d6] Submitted process > SAMTOOLS_INDEX (2)
[18/21a0ae] Submitted process > GATK_HAPLOTYPECALLER (1)
[f0/4e8155] Submitted process > GATK_HAPLOTYPECALLER (2)
[d5/73e1c4] Submitted process > GATK_HAPLOTYPECALLER (3)
Takeaway¶
You know how to make a variant calling workflow handle a basic samplesheet.
What's next?¶
Add a joint genotyping step that combines the data from all the samples.
6. Stretch goal: Add joint genotyping step¶
To complicate matters a little, the GATK variant calling method calls for a consolidation step where we combine and re-analyze the variant calls obtained per sample in order to obtain definitive 'joint' variant calls for a group or cohort of samples (in this case, the family trio).

This involves using a GATK tool called GenomicsDBImport that combines the per-sample calls into a sort of mini-database, followed by another GATK tool, GenotypeGVCFs, which performs the actual 'joint genotyping' analysis. These two tools can be run in series within the same process.
One slight complication is that these tools require the use of a sample map that lists per-sample GVCF files, which is different enough from a samplesheet that we need to generate it separately. And for that, we need to pass the sample ID between processes.
Tip
For a more sophisticated and efficient method of metadata propagation, see the topic of meta maps.
6.2. Add the sample ID to the tuple emitted by SAMTOOLS_INDEX¶
Before:
After:
6.3. Add the sample ID to the GATK_HAPLOTYPECALLER process input and output definitions¶
Before:
input:
tuple path(input_bam), path(input_bam_index)
...
output:
path "${input_bam}.g.vcf"
path "${input_bam}.g.vcf.idx"
After:
input:
tuple val(id), path(input_bam), path(input_bam_index)
...
output:
tuple val(id), path("${input_bam}.g.vcf"), path("${input_bam}.g.vcf.idx")
6.4. Generate a sample map based on the output of GATK_HAPLOTYPECALLER¶
// Create a sample map of the output GVCFs
sample_map = GATK_HAPLOTYPECALLER.out.collectFile(){ id, gvcf, idx ->
["${params.cohort_name}_map.tsv", "${id}\t${gvcf}\t${idx}\n"]
}
6.5. Write a process that wraps GenomicsDBImport and GenotypeGVCFs called GATK_JOINTGENOTYPING¶
/*
* Consolidate GVCFs and apply joint genotyping analysis
*/
process GATK_JOINTGENOTYPING {
container "broadinstitute/gatk:4.5.0.0"
input:
path(sample_map)
val(cohort_name)
path ref_fasta
path ref_index
path ref_dict
path interval_list
output:
path "${cohort_name}.joint.vcf"
path "${cohort_name}.joint.vcf.idx"
"""
gatk GenomicsDBImport \
--sample-name-map ${sample_map} \
--genomicsdb-workspace-path ${cohort_name}_gdb \
-L ${interval_list}
gatk GenotypeGVCFs \
-R ${ref_fasta} \
-V gendb://${cohort_name}_gdb \
-O ${cohort_name}.joint.vcf \
-L ${interval_list}
"""
}
6.6. Add call to workflow block to run GATK_JOINTGENOTYPING¶
// Consolidate GVCFs and apply joint genotyping analysis
GATK_JOINTGENOTYPING(
sample_map,
params.cohort_name,
params.genome_reference,
params.genome_reference_index,
params.genome_reference_dict,
params.calling_intervals
)
6.7. Add default value for the cohort name parameter up top¶
6.8. Run the workflow to verify that it generates the final VCF output as expected¶
Now we see the additional process show up in the log output (showing the compact view):
N E X T F L O W ~ version 23.10.1
Launching `hello-gatk.nf` [nauseous_thompson] DSL2 - revision: b346a53aae
executor > local (7)
[d1/43979a] process > SAMTOOLS_INDEX (2) [100%] 3 of 3 ✔
[20/247592] process > GATK_HAPLOTYPECALLER (3) [100%] 3 of 3 ✔
[14/7145b6] process > GATK_JOINTGENOTYPING (1) [100%] 1 of 1 ✔
You can find the final output file, family_trio.joint.vcf, in the work directory for the last process. Click on it to open it and you'll see 40 lines of metadata header followed by just under 30 jointly genotyped variant records (meaning at least one of the family members has a variant genotype at each genomic position listed).
Tip
Keep in mind the data files covered only a tiny portion of chromosome 20; the real size of a variant callset would be counted in millions of variants. That's why we use only tiny subsets of data for training purposes!
Takeaway¶
You know how to make a joint variant calling workflow that outputs a cohort VCF.
What's next?¶
Celebrate your success and take an extra long break! This was tough and you deserve it.
In future trainings, you'll learn more sophisticated methods for managing inputs and outputs (including using the publishDir directive to save the outputs you care about to a storage directory).
Good luck!